The second state of the art lecture at the 2022 BSTH annual meeting focussed on atherothrombosis. In a first lecture of this session, Prof. Lina Badimon (Hospital de la Santa Crue and Sant Pau, Barcelona, Spain) addressed the recent progress in antiplatelet therapy and the different factors that contribute to the formation of thrombi. Thereafter, Dr. Matthias Engelen (University Leuven) discussed antithrombotic therapy in COVID-19 and provided an overview of the updated BSTH guidelines in this field. Finally, Dr. Marie-Astrid Van Dievoet, (Cliniques Universitaires Saint-Luc, Brussels, Belgium) closed the session with a presentation on platelets in chronic liver disease as well as preliminary results from her own research work in this setting.
Presented by: Lina Badimon, MD, PhD (Hospital de la Santa Crue and Sant Pau, Barcelona, Spain)
Since the introduction of the first pharmacological therapy for the treatment of patients with acute myocardial infarction in the early 20th century, there has been a steep decline in mortality and morbidity in patients with ST-segment elevation myocardial infarction (STEMI).1 However, since 2010, we have again witnessed a slight increase in cardiovascular disease mortality. This can potentially be explained by the aging population in combination with the growing incidence of obesity among the population.2 Interestingly, these trends hold true for all regions of the world, including both high- and low-income countries.3 To date, cardiovascular diseases (CVDs), which include coronary heart disease, hypertension, and stroke, collectively comprise the number one cause of death globally.4
The thrombogenicity of a disrupted atherosclerotic lesion depends on the nature and extent of the plaque components exposed to flowing blood, together with local rheology and a variety of systemic factors. Previously, studies have reported differences in thrombogenicity of various types of human atherosclerotic lesions when exposed to flowing blood in a well-characterised perfusion system. In this, the atheromatous core is the most thrombogenic component of human atherosclerotic plaques. Subsequently, research focussed on the role of tissue factor (TF) in the thrombogenicity of different types of plaques was investigated. TF is present in lipid-rich human atherosclerotic plaques and is most likely an important determinant of the thrombogenicity of human atherosclerotic lesions after plaque disruption. This TF activity can however be inhibited through TF pathway inhibition. The latter is of particular clinica; relevance since TF is not present in the intima of healthy vessels but only appears in the context of a vessel break. To date, however, there is no drug that specifically targets this pathway and further studies are needed.5-7 In addition to that, TF also regulates microvessel formation and stabilisation by induction of chemokine ligand 2 expression.8
High-risk plaques are characterized by a large lipid core, a vulnerable necrotic core, a thin fibrous cap, an infiltrate of innate and adaptive immune cells and lots of inflammation. In contrast, they have less smooth muscle cells and have a lack of supporting collagen fibres. This combination can trigger a structural failure due to plaque erosion or disruption, after which the thrombus can start to grow on the surface. Haemostatic plug formation, which usually occurs at very high blood shear rates, and thrombosis on endothelial injured vessels lead to rapid platelet recruitment. The initial tethering is mainly mediated by the interaction between platelet glycoprotein (GP)Ibα receptor and the A1 domain of von Willebrand factor (vWF). In contrast, in atherosclerotic plaques, the initial trigger is exposed TF whoch induces thrombin and the subsequent formation of a fibrin monolayer covering the surface of the exposed vascular damage. Later, thrombosis evolves with a predominance of platelets that are rapidly activated and recruited to the growing thrombus.9
Antithrombotic therapy and risk stratification
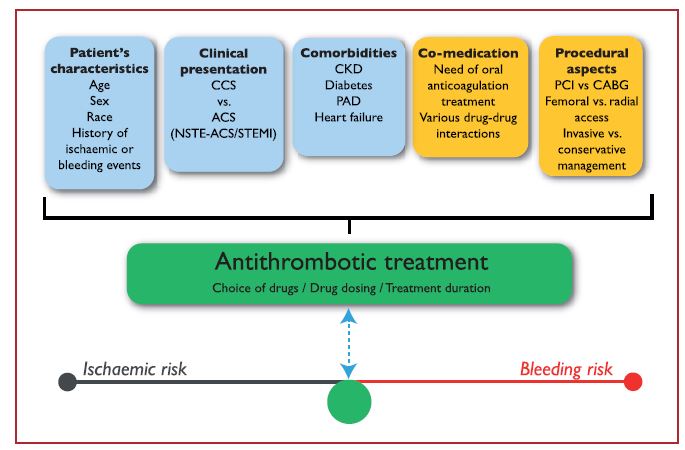
The choice, combination, timing of treatment initiation and treatment duration of antithrombotic therapy all depend on various intrinsic and extrinsic factors (Figure 1). Importantly, while CVD patients were previously only treated with heparins, antiplatelet therapy can now also be used. To date, the most commonly used antiplatelet targets to prevent cardiovascular events consist of ADP receptor antagonists and thromboxane pathway inhibitors. Especially the P2Y12 inhibitors are gaining momentum. Other drug classes that are under investigation in this setting include thromboxane receptor blockers, PDE inhibitors, PAR inhibitors and GPIIb/IIIa inhibitors.10,11
Figure 1. Determinants of antithrombotic treatment in coronary artery disease.11
Dynamic process
Thrombus formation is a dynamic process. To learn more about the changes in thrombus composition in acute myocardial infarction, a study was performed to investigate the characteristics of human coronary thrombus in STEMI. In this study, intracoronary thrombi and blood from the culprit coronary site and the systemic circulation were obtained during percutaneous coronary intervention (PCI). Thrombi were categorised by onset-of-pain-to-PCI elapsed time in thrombus of <3 (T3) and more than 6 hours of evolution (T6). While T3 thrombi were mainly composed by platelets and fibrin(ogen), T6 thrombi were characterised by a reduced platelet content, an increased level of leukocyte infiltration (including monocytes, neutrophils, T-cells, and B-cells), and the appearance of undifferentiated progenitor cells. Confocal microscopy showed that undifferentiated CD105+ cells were absent in T3, but did infiltrate the T6 coronary thrombi. Immunohistochemical analysis confirmed the presence of undifferentiated CD34+ and CD105+ cells in aged occlusive thrombi.12
Another component adding to the complexity of thrombosis in coronary artery disease are extracellular vesicles. Indeed, platelet activation induced by thrombin triggers the shedding of platelet-released extracellular vesicles with a complex proteomic pattern rich in procoagulant and pro-adhesive proteins.13 Furthermore, also platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques.14 Finally, it was shown that while serum native C-reactive protein (CRP) may not affect thrombus growth, monomeric CRP does have a prothombotic effect, enhancing not only platelet disposition but also thrombus growth under arterial flow conditions.15
Novel targets and platelet inhibitors
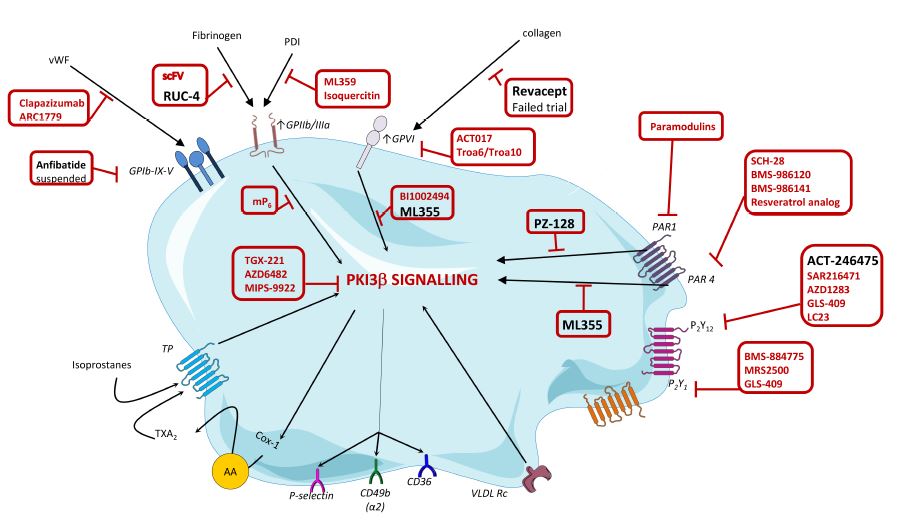
Over the last years, the search for new potent and safer antiplatelet agents has continued at a steady pace and several drugs have already reached clinical development (drugs depicted in black in Figure 2). One of these drugs is RUC-4 which potently interferes with the binding of fibrinogen to GPIIb/IIIa. In addition, also the novel GPVI antagonist revacept has been tested in a phase II clinical trial but failed to show any benefit. However, many other agents are still in the pipeline. Among others, this includes ML355, a compound that blocks AKT, PI3K and Erk1/2 and PZ-128, a first-in-class cell penetrating lipopeptide pepducin which inhibits PAR-1-G protein signalling pathway by targeting the intracellular surface of the receptor. Finally, selatogrel is a novel, highly selective P2Y12 inhibitor. In the years to come we will find out if and how these agents survive scrutiny in clinical trials and actually make it to daily clinical practice.1
Figure 2. Targets and novel platelet inhibitor molecules.1
Conclusion
In coronary artery disease, many factors contribute to the formation of thrombi. First, there are many systemic factors, including lipoproteins, diabetes, obesity, smoking and hypertension, which lead to high thrombogenicity. In addition, extracellular microvesicles and inflammation contribute to thrombus formation. Secondly, local factors such as fluid dynamics (shear stress) and the nature of the exposed substrate (erosion or rupture, atherosclerotic plaque composition, etc.) play a major role. Although all these factors contribute to a very complex disease biology, it also opens up the path towards many new therapeutic strategies.
Presented by: Matthias Engelen, MD (University Leuven)
Over the past three years, COVID-19 posed extreme challenges to the medical community. Especially during the early days the of the pandemic, we have witnessed a high rate of mortality, mainly due to an overwhelmed healthcare system and a lack of disease-modifying drugs. By now, it is well-established that COVID-19 is frequently associated with thrombotic events, including major thrombotic events and diffuse activation of coagulation and the formation of microthrombi. Despite usual doses of thromboprophylaxis, there was a high rate of venous thromboembolism (VTE) in hospitalised patients with COVID-19, mainly among patients admitted to the intensive care unit (ICU).16 Furthermore, when patients presented with high D-dimer levels (>2 µg/ml at admission), there was a 50-fold higher mortality rate.17 In order to improve clinical outcomes, clinicians sought to prevent thrombosis. Indeed, early observational studies at the beginning of the pandemic demonstrated a decreased mortality in patients treated with anticoagulation. Potential benefits of anticoagulation or heparin resulted in a reduction of thrombosis, a reduction in systemic inflammation and perhaps even some antiviral effects. However, there were no randomised controlled trials available. To address this issue, the BSTH composed a guidance document on the treatment and prevention of VTE, mostly based on expert opinions.
By now, the results of many randomised trials in the COVID-19 setting have been published. However, the COVID-19 era did form a very challenging timeframe to conduct good clinical studies all studies. Indeed, different analytical approaches were used across the various studies with diverging primary endpoints, making comparison very difficult. In addition, the virus continues to evolve over time. As a result, some of the trials seemingly produced conflicting evidence on the optimal anticoagulation strategy in these patients. Therefore, a meta-analysis including 11 randomised controlled trials compared intensified vs. prophylactic anticoagulation in patients with COVID-19. Intensified vs. prophylactic anticoagulation was not associated with a mortality reduction up to 45 days. However, the risk of VTE was reduced, with effect driven by therapeutic rather than intermediate dosing, while major bleeding was increased with intensified anticoagulation (even though patients with risk factors for bleeding were almost always excluded from participation).18
Updated BSTH guidelines
COVID-19 outpatient
For COVID-19 outpatients with a new or known VTE and/or prior indications for therapeutic anticoagulation (tAC), tAC following the conventional guidelines should be administered. In patients without new or known VTE and without indication for tAC, there is no need for routine prophylaxis. However, one must remain vigilant for signs of thrombosis. In addition, patients should also be reassessed on a regular basis and when hospitalisation is required, the guidelines for hospitalised patients (see below) need to be followed.
Hospitalised COVID-19 patient
In the hospitalised setting, there are three possible dosing regimens of low molecular weight heparins (LMWH), all of which are weight adjusted. In the prophylactic dose of LMWH (pLMWH), heparins should be administered at 50 IU/kg once daily. For the intermediate regimen (iLMWH), 50 IU/kg are administered twice daily (once daily in patients with renal insufficiency) while therapeutic dosages (tLMWH) consist of 100 IU/kg twice daily (once daily in patients with renal insufficiency). For hospitalised patients, three main questions need to be answered: (I) Is there an indication for tAC? (II) Is there a need for VTE diagnostics? (III) What is the bleeding risk?
When a new VTE is diagnosed, tLMWH should be initiated. If there was a prior indication for anticoagulation, oral anticoagulation can be continued when the patient is asymptomatic and when there is a good oral bioavailability. When these factors cannot be guaranteed, one needs to switch to tLMWH. For patients without a prior indication for anticoagulation and no known VTE, the new BSTH guidelines recommend to opt for prophylactic doses of LMWH, both for ward and ICU patients. However, in selected ward patients (i.e., patients with a low bleeding risk, high D-dimers and low-flow oxygen) therapeutic doses of LMWH can be considered. In contrast, these therapeutic doses of LMWH are generally not recommended in ICU patients. Nonetheless, some trials do show a reduction in the thrombosis risk in patients with low bleeding risk, fibrinogen levels that are above 200 mg/dL, no dual antiplatelet therapy (DAPT) and no recent ischemic stroke. However, as the bleeding risk in these patients, despite all these criteria, increases significantly, tLMWH is not recommended in ICU patients.
With respect to iLMWH in ICU patients, guidelines are less clear. A Belgian study therefore analysed the VTE incidence and bleeding before and after implementing a hospital-wide intensified thromboprophylaxis protocol (2020 BSTH guidelines) in 412 patients with COVID-19. In hospitalised patients with COVID-19, there were no additional symptomatic VTEs and a reduction in incidental deep vein thrombosis after implementing systematic thromboprophylaxis with weight-adjusted prophylactic (ward) to intermediate (ICU), but not therapeutic dosed anticoagulation. This intensified thromboprophylaxis was associated with a lower risk of major bleeding compared with therapeutically dosed anticoagulation (1.2% vs. 7.7%).19 Furthermore, there are also randomised trials comparing intermediate and standard-dose prophylactic LMWH in critically ill COVID-19 patients. These trials show that there is no difference in the prevention of death, nor in major bleeding rate.20,21 Therefore, also intermediate doses of LMWH can be considered as good clinical practice. Importantly, the patient and his/her indications for therapeutic anticoagulation, VTE diagnostics and bleeding risk should be reassessed frequently.
Discharged COVID-19 patient
If the patient is ready for discharge, patients with a new diagnosis of VTE or a prior indication for anticoagulation should follow the conventional guidelines. For the other patients, risk factors for VTE should be assessed. When at discharge patients have an IMPROVEDD score of ≥4 or an IMPROVE score of ≥2, no bleeding risk, no DAPT and a creatinine clearance above 30 ml/min, post-discharge prophylactic anticoagulation can be considered. When there is no VTE risk, patients should not receive post-discharge thromboprophylaxis.
Conclusion
COVID-19 is associated with an increased risk for thrombotic complications. Available clinical trials assessing thromboprophylaxis seemingly produce conflicting evidence due to the burdensome circumstances, divergent endpoints, different analytic approaches and the evolving COVID-19 disease. All these factors hamper a comparison and extrapolation of available evidence. Therefore, the BSTH is working on updated guidance to provide thromboprophylaxis in COVID-19 patients. Clinicians should provide thromboprophylaxis in hospitalised COVID-19 patients while (re)assessing bleeding and thrombotic risk frequently.
Presented by: Marie-Astrid Van Dievoet, MD (Cliniques Universitaires Saint-Luc, Brussels, Belgium)
Chronic liver disease and cirrhosis is a very heterogeneous disease that can present in both children and adults with different aetiologies, varying levels of severity (e.g., compensated or decompensated). Patients with chronic liver disease are believed to have a rebalanced haemostatic system in the three phases of haemostasis; primary haemostasis, the coagulation cascade and fibrinolysis. For example, in primary haemostasis there is thrombocytopenia as an anticoagulant driver promoting bleeding, while this is counterbalanced by elevated levels of Von Willebrand factor, promoting thrombosis. In the coagulation cascade, there is a decrease in coagulation factors II, V, VII, IX, X, XI, XII (promoting bleeding) while on the other hand, there is a low level of antithrombin, protein C and protein S (promoting thrombosis). Finally, in fibrinolysis, there are high levels of tissue plasminogen activator but low levels of plasminogen. It is to no surprise that this balance is very fragile and may tip to (haemostasis-related) bleeding or thrombosis.22
The majority of patients with chronic liver disease has thrombocytopenia, although usually mild or moderate. This thrombocytopenia is multifactorial and can be explained by a decreased platelet production and a decreased platelet survival. To date, there are conflicting reports on the link between platelet count and procedural bleeding risk.23-25 Furthermore, research on platelet function is not yet converging. Several papers show platelet hypofunction in patients with liver cirrhosis, while other studies point towards a normal or enhanced platelet function. As such, these lines of evidence or not yet conclusive.26-32 In terms of haemostasis, a preserved primary haemostasis was observed in vitro. In flow based assays a normal to enhanced platelet adhesion was observed, probably due to high plasma levels of Von Willebrand Factor and low ADAMTS13 activity.33,34 Furthermore, also some clinical observations suggest preserved primary haemostasis. In this, liver transplantation is possible without correction of the platelet count, patients are not protected from arterial/venous thrombosis and there is an increased thrombosis risk with thrombopoietin-receptor agonists.35-37
To better address this issue, Dr. Van Dievoet is conducting a descriptive clinical study of haemostasis in decompensated children and adults. The main focus of the study are platelets, but the different three phases of the haemostatic system as a whole are also being studied. In the trial, blood samples before and after hepatic transplantation are taken and the evolution of platelets is measured. In this, procoagulant platelets are measured with two different techniques, the Maastricht flow chamber and by flow cytometry. In a flow cytometry experiment, phosphatidylserine exposure after maximal stimulation seems to be lower in cirrhotic children than in healthy controls. Furthermore, the phosphatidylserine exposure in paediatric patients seems to differ from controls depending on the microspot that was used in the flow chamber experiment. The mechanism behind this effect is not yet known and is currently being explored.
Conclusion
In cirrhotic patients there is a relatively preserved primary haemostasis. Thus far, studies on the cirrhotic platelet phenotype are scarce and inconclusive. Researchers believe that flow-based and flow cytometry assays could enhance our understanding of this topic. Although preliminary results are promising, they need further investigation.
References
