The first state of the art lecture at the 2022 BSTH annual meeting focussed on platelet research. In a first lecture of this session, Prof. Jonathan Douxfils (University of Namur, Belgium) addressed the interplay between platelets and coagulation, and explained how further research into this interaction may facilitate a better understanding of complex bleeding disorders. Prof. Robert Ahrends (University of Vienna, Austria) followed up with a lecture onhow lipidomics can be used to unravel platelet function and reveal therapeutic opportunities. Finally, Prof. Claudia Tersteeg (KU Leuven, Belgium) wrapped up the session with a presentation on the role of procoagulant platelets in controlling normal haemostasis and mediate thrombosis and thrombo-inflammation.
Despite being highly interconnected, coagulation and platelets have mainly been analysed individually for many years. For the analysis of platelet function, the main principles for testing include turbidimetric-based optical detection methods in platelet-rich plasma (PRP) or whole blood, changes in electrical impedance between electrodes in whole blood, time to thrombus formation in high-shear blood flow through a collagen-coated membrane using whole blood, quantification of platelet adherence to a plate under arterial flow conditions using whole blood and the use of fluorophore-labelled antibodies against surface glycoproteins and surface-bound activating markers. Some of the tests that are routinely being used consist of a platelet aggregation profiler (PAP4 or PAP8), a chronology/multiplate analyser, a platelet function analyser (PFA 100/200), VerifyNow, IMPACT (Cone and Plate[Let] analyser), PateletWorks, Flow cytometry, and ROTEM or TEG platelet mapping.
When studying coagulation, the main principles for testing also include turbidimetric-based optical detection in platelet-poor plasma (PPP), PRP or whole blood, mechanical clot detection in PPP, PRP or whole blood, visco-elastometric methods, sonic estimation of elasticity via resonance, fluorometric monitoring of a particular enzyme activity and visual monitoring of clot formation. The most commonly used coagulation tests are TEG, ROTEM, thrombin generation, clot waveform analysis, overall haemostatic potential, FibWave, sonorheometry, thrombodynamics. Although these tests can provide useful information, they don’t capture the interplay between coagulation and platelets.1 During haemostasis, the formation of prothrombin and prothrombinase complexes starts the amplification phase of coagulation. Later, prothrombin transforms into thrombin, which in turn leads to platelet activation. Activated platelets expose specific phospholipids to the surface and release activated factors V and VIII which initiates the propagation phase of the coagulation cascade.1,2 Of note, thrombin is able to activate platelets, but platelets are also able to release factors that can downregulate thrombin formation.3 As such, platelets and coagulation are highly interconnected, and this interaction cannot be assessed with the current coagulation tests, in which phospholipids are added to assess the transformation of fibrinogen into fibrin.
However, some of the available tests do allow us to assess this interplay. This includes visco-elastometric detection of clot formation, turbidimetric measurement in PRP and thrombin generation in PRP or whole blood.4 Capturing this interplay is important for several reasons. For example, direct oral anticoagulants (DOACs) can affect platelet function testing. In fact, it has been observed that there is a reduction in platelet aggregation two hours after the intake of edoxaban. This means that, despite the fact that there is no direct impact on platelet function, edoxaban does have an indirect impact via the coagulation cascade. In contrast, platelet aggregation increases in patients taking dabigatran, which can be explained by an increased production of the thrombin receptor activating peptide (TRAP) by platelets.5,6 Another example illustrating the importance of a better understanding of the platelet-coagulation pathways is that commonly encountered platelet function disorders are associated with abnormalities in thrombin generation. Patients with platelet function disorders have a particular thrombin generation pattern that correlates with clinical events (e.g., wound healing problems, minor wound bleeding lasting >1 hour). This suggests that thrombin generation patterns may be related to the bleeding tendency in these patients.7 A special analysis method that can be used in this context consists of the calibrated automated thrombogram (CAT), in which platelets can aggregate before putting them into the thrombin generation test.8 Using this method, Didelot et al. were able to see very different patterns with activated and aggregated PRP, meaning that activation of platelets can occur both in vitro and in vivo. They also revealed different patterns in thrombin-generation parameters, meaning that, by using different inducers or cocktails of inducers, both the contribution of platelets and the status of the coagulation cascade could be analysed.1
In conclusion, evaluating the interplay between platelets and coagulation may increase our understanding of complex bleeding disorders. The techniques for such an evaluation are already available, but a proper assessment of their added benefit is required, and new inducers and reagents need to be developed. This concomitant evaluation is not unrealistic but it does require standardisation and further validation.1
Lipidomics refers to “the emerging scientific discipline of studying the full lipid complement of cells, tissues, and organisms (K. Simon)”. Lipids are involved in variety wide range of processes, including cell migration, transcriptional control, transport, stress response, inflammation and clotting (via the formation of vesicles). The function of lipids derive from their structure. In membranes, of which lipids constitute a significant part, lipids with a very polar head and a long tail cannot flip. In contrast, phospholipids, with two chains and a medium polar head group, can easily diffuse in membranes and can be transported, making them perfect vehicles for signal transduction. It is important to know that lipids are not just floating around the membrane. In fact, they can form very ordered structures, such as lipid rafts in which myelins and ceramides can be found. These platforms play an important role important for interactions with the cytoskeleton, and for the functioning of membrane receptors and channels. From a metabolomics point of view, lipids make up the largest group of molecules in the human body. Some of the challenges related to lipidomics research are biological in nature: there is a high diversity of lipids (> 40,000 species), they have a varying stability (some are more stable than others), come with a huge concentration range (109), and are subject to spatial redundancy and a high turnover (much faster than proteins). From a technical point of view, the novelty of this research area means that there is a lack of reference material and standards. The biggest challenge, however, lies in the lipid diversity, including different backbones, bonds (for example, esther, ether bonds), the number of hydroxyl-groups and variations in head groups. Due to its huge variety, different spectrometry and separating techniques are needed to cover the entire range.
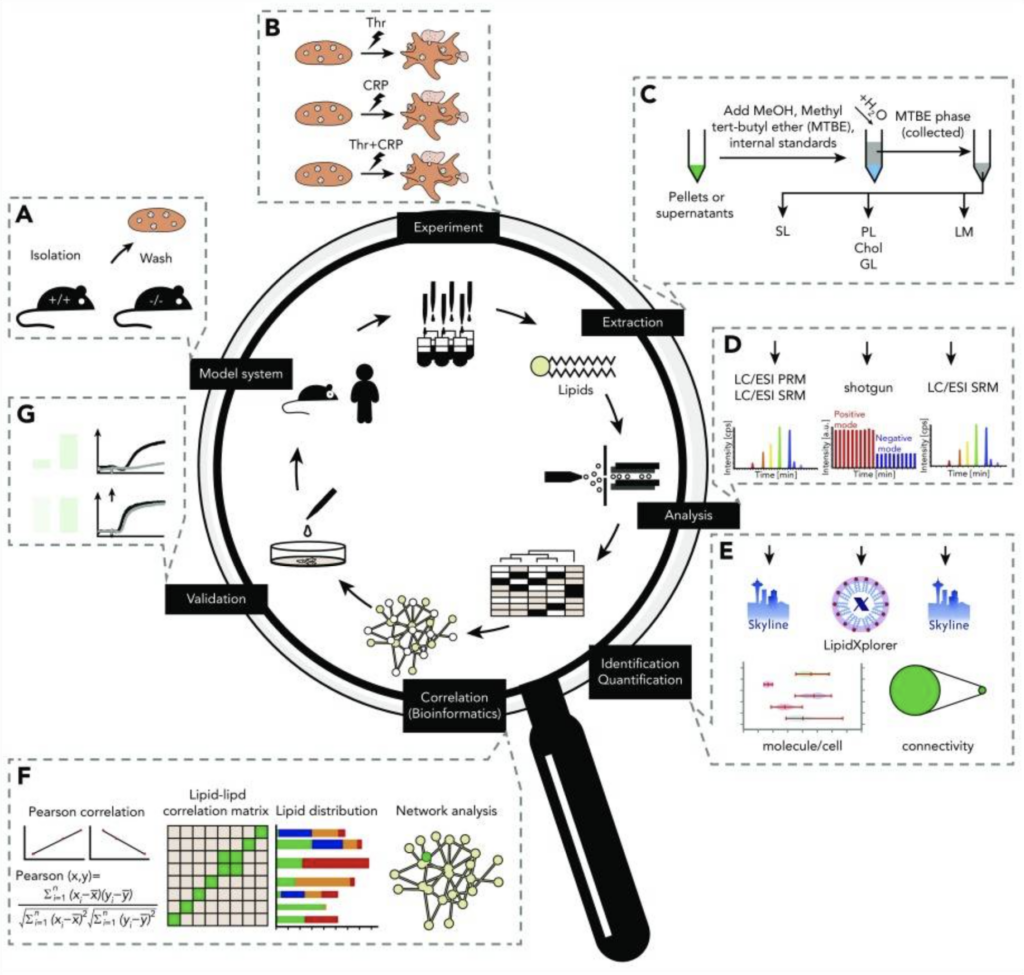
Specifically in platelets, lipids are involved in membrane transformation and signalling (lipids are involved in at least six different signalling pathways). In the group of Dr. Ahrends, a comprehensive platelet lipidomics platform is used to analyse these platelet-related lipids (Figure 1). In brief, blood cells (mice or human) are first isolated after which platelets are stimulated. Subsequently, the different molecular lipid classes are extracted (membrane fractions), which are later analysed by membrane lipidomics (LC/ESI PRM, LC/ESI SRM and shotgun) or signalling lipidomics (LC/NSI SRM and IC/ESI SRM). After this, lipids are identified and quantified, followed by data integration (network analysis and database) and validation. This platform allows researchers to extract information on membrane properties, lipid signalling and energy consumption or storage. It also allows the extraction of information about the structure of the lipids (number and type of bonds, type of fatty acids, etc.), which is a key objective for Dr. Ahrends and his group. This technique covers around 500 lipids over eight-quarters orders of magnitude. Mouse and human lipidomics are similar in dynamic range. Notably, only 15 lipids account for up to 70% of the entire platelet lipid mass, including a high amount of PUFA lipids that are important for signalling in platelets. A systematic comparison of the lipidomics network in resting and activated murine platelets, validated in human platelets, revealed that <20% of the platelet lipidome is changed upon activation, involving mainly lipids containing arachidonic acid (AA). Of note, PI 18:0-20:4 is the major precursor for the production of AA. This means that murine and human platelet lipidomes are generally stable and that only a set of key lipids change upon activation. Interestingly, this platform also allows investigators to study certain lipids that are relevant for specific diseases. As a proof of concept, SMPD1– deficient mice were used. Sphingomyelin phosphodiesterase (SMPD1) controls the equilibrium between sphingomyelin and ceramide, and SMPD1-deficiency results in a very specific modulation of the platelet lipidome. While most of the lipidome remains stable, lyso-sphingomyelin (SPC) is upregulated10 fold, with subsequent modification of platelet aggregation. This sheds more light on novel mechanisms important for platelet function, and has, therefore, the potential to open novel diagnostic and therapeutic opportunities.9
In conclusion, it is important to remember that the platelet lipidome displays a huge dynamic range. Only 15 lipids make up to 70% of the platelet lipidome in resting platelets and less than 20% of the platelet lipidome is changed upon activation. This mainly consists of lipids containing AA. Using SMPD1-deficient mice, SPC was shown to have a function in platelet aggregation, opening the door to a future in which lipidomics can help to gather more information on platelet function in disease models.9
Figure 1. Comprehensive platelet lipidomics platform.9
The aggregation of platelets and formation of the platelet plug is essential to stop the bleeding. These activated platelets that form the plug have been broadly studied. However, the plug also contains procoagulant platelets. These platelets expose phosphatidylserine (PS) on their surface, which facilitates the assembly of the prothrombinase complex and, therefore, the transformation of prothrombin to thrombin that results in the formation of fibrin.10,11 The way in which these procoagulant platelets is formed is still a matter of debate. In vitro, the addition of colvulxin and thrombin results in the activation of platelets. All platelets in the solution become activated, and there is an increase in the levels of cytosolic Ca2+ and activation of integrin aIIbb3, which is essential for the aggregation of platelets. In about two minutes, two populations of platelets can be distinguished: an aggregating and a procoagulant type. In aggregating platelets, Ca2+ levels slightly decrease after differentiation, and they express integrin aIIbb3 on their surface. In procoagulant platelets, the cytosolic Ca2+ level increases, which seems to be essential in the formation of these procoagulant platelets. This Ca2+ level increase eventually results in mitochondrial failure and inactivation of integrin aIIbb3. As a result, these platelets are no longer able to take part in the aggregate. Furthermore, these platelets lose membrane integrity, expose PS, have a coat of a-granules on their surface, and ultimately start to balloon and fragment.12 However, there remain to be many unanswered questions on this process: Which intracellular signalling cascades are involved?; Why is it that only a subset of platelets becomes procoagulant?; What is the clinical relevance of procoagulant platelet formation?; etc. A more detailed analysis of the process shows that, in procoagulant platelets, the increase in cytosolic Ca2+ levels is a result of a reversal of the Ca2+/Na+ exchange, leading to an increased influx of calcium into platelet instead of Ca2+ efflux. The mitochondria incorporate this intracellular Ca2+ until the level becomes too high. At this moment, the mitochondrial permeability transition pore (mPTP) is formed, which depends on cyclophilin D (CypD). Through this pore, Ca2+ leaves the mitochondria, resulting in a loss of mitochondrial membrane potential and ATP depletion, eventually leading to mitochondrial failure. When Ca2+ leaves the mitochondria, this activates TMEM16F, which brings PS from the inside of the membrane to the outside. This PS exposure leads to an activation of the prothrombinase complex and fibrin formation.13,14 This process is also associated with the formation of very long platelet protrusions, which will eventually break and form microparticles.15
To figure out why some platelets become procoagulant, the group of Dr. Tersteeg are currently focussing on SIRT3, which is a major deacetylase in mitochondria. SIRT3 can deacetylate CypD, which increases the threshold for mPTP formation and therefore protects the cell from necrosis. It is expected that SIRT3 plays a similar role in platelets. SIRT3 has been shown to be present in both mouse and human platelets. In SIRT3-deficient mice, activation and aggregation of platelets is not affected, but these mice did have a much faster clot formation in vivo.16 The underlying hypothesis for this is that the absence of SIRT3 results in an increased presence of procoagulant platelets. This hypothesis has been validated in a recent experiment (van Bael et al., in preparation). Patients with Scott syndrome have a mutation in TMEM16F, so their procoagulant platelets cannot expose PS on the outer surface. This results in a mild bleeding disorder.17,18 The same phenotype was demonstrated in CypD-deficient mice. As such, this demonstrates that PS is necessary for coagulation. In thrombosis models there are, however, conflicting data. A study showed that there is an accelerated occlusion time in CypD-deficient mice. The authors suggested that this accelerated occlusion time occurs because integrin aIIbb3 cannot be inactivated leading to an inability to form procoagulant platelets, with the formation of pro-aggregating platelets instead.19 Others, however, have shown the opposite effect in which a deficiency in CypD results in a decreased thrombus size and fibrin formation.20
Procoagulant platelets also seem to have a function in thrombo-inflammation. It has been shown that neutrophils can interact with the long prolongations of procoagulant platelets, which results in neutrophil activation and neutrophil-platelet aggregation.15 In ischemia models, Yuan et al. observed that neutrophil aggregation is enhanced in the presence of PS-positive platelets.21 Similar observations were made by Denorme et al., showing that mice with ischemia had an increased number of circulating procoagulant platelets with a tendency to bind neutrophils.22 In contrast, a lack of procoagulant platelets in CypD-deficient mice resulted in inhibition of both neutrophil aggregation and thrombi formation. Therefore, CypD-deficient mice were found to be protected from ischemia-reperfusion (IR) injuries.21,22 As such, this suggests that procoagulant platelets exacerbate ischemia-reperfusion (IR) injury. However, a very recent study showed that procoagulant platelets were able to maintain vascular integrity. Activation of procoagulant platelets promotes binding of the prothrombinase complex to the platelet membrane, greatly enhancing thrombin activity and resulting in fibrin formation, which results in protection from bleeding.23 In CypD-deficient mice, investigators observed fewer procoagulant platelets and more inflammatory bleeding. As such, the benefits/drawbacks of procoagulant platelets are model dependent. They can enhance thrombo-inflammation, but on the other hand, they may also help to prevent bleeding. Based on these observations, procoagulant platelet inhibition could be a novel therapeutic target. Specific inhibition could be aquaporin inhibitors, increased flippase activity to reduce PS exposure and CytD inhibition.24,25
In conclusion, more research is needed to understand underlying molecular mechanisms and the precise function of procoagulant platelets in vivo. However, we already know that PS exposure in procoagulant platelets is required for normal haemostasis. Procoagulant platelets mediate thrombosis and thrombo-inflammation. Therefore, inhibition of these platelets might be a novel target to prevent these problems.10
References
1. Douxfils J. Presented at BSTH 2022.
2. Kubitza D and Haas S. Expert Opin Investig Drugs. 2006;15(8):843-55.
3. Swieringa F, et al. Res Pract Thromb Haemost. 2018;2(3):450-60.
4. Evrard J, et al. Res Pract Thromb Haemost. 2022;6(7).
5. Olivier CB, et al. Thromb Res. 2016;138:63-8.
6. Sokol J, et al. J Thromb Thrombolysis. 2018;46(3):393-8.
7. Sharma T, et al. Int J Lab Hematol. 2021;43(6):1557-65.
8. Didelot M, et al. Platelets. 2018;29(2):156-61.
9. Ahrends R. et al. Presented at BSTH 2022.
10. Tersteeg C. Presented at BSTH 2022.
11. van der Meijden PE and Heemskerk JW. Nat Rev Cardiol. 2019;16(3):166-79.
12. Aliotta A, et al. Thromb Haemost. 2021;121(3):309-21.
13. Veuthey L, et al. Int J Mol Sci. 2022;23(5).
14. Yang H, et al. Cell. 2012;151(1):111-22.
15. Tersteeg C, et al. Circ Res. 2014;114(5):780-91.
16. Gaul DS, et al. Cardiovasc Res. 2018;114(8):1178-88.
17. Suzuki J, et al. Nature. 2010;468(7325):834-40.
18. Baig AA, et al. Arterioscler Thromb Vasc Biol. 2016;36(11):2152-7.
19. Jobe SM, et al. Blood. 2008;111(3):1257-65.
20. Hua VM, et al. Blood. 2015;126(26):2852-62.
21. Yuan Y, et al. Sci Transl Med. 2017;9(409).
22. Denorme F, et al. Blood. 2020;135(6):429-40.
23. Kaiser R, et al. Blood. 2022;140(2):121-39.
24. Agbani E et al.. Presented at ISTH 2017.
25. Millington-Burgess SL and Harper MT. J Thromb Haemost. 2022;20(4):989-995.
