During the educational session of the annual BSTH meeting, Prof. Katrien Devreese (Ghent University Hospital, Ghent, Belgium) gave an overview on how to adequately diagnose acquired thrombotic thrombocytopenic purpura (TTP). In this, an ADAMTS13 activity below 10% (10 IU/dL) is diagnostic in patients with a clinical scenario of TTP. To date, several assays are available to determine ADAMTS13 activity, including functional FRET based assays, chromogenic ELISA assays and rapid screenings. Unfortunately, test results are not always readily available, which may cause problems in emergency situations. Apart from ADAMTS13 activity measurements, also an ADAMTS13 antibody assessment is necessary to discriminate between congenital or immune-mediated TTP. Finally, ADAMTS13 activity should be monitored throughout TTP treatment in order to timely detect the risk of exacerbation or relapse.
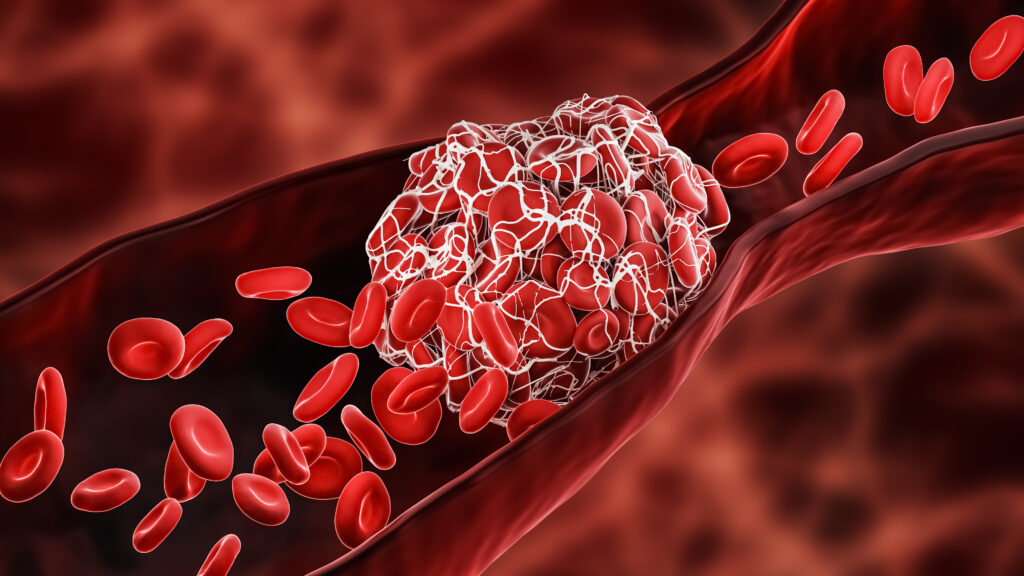
Thrombotic thrombocytopenic purpura (TTP) was first described in 1925 in a case-report of a 16-year old girl presenting with fever, haemolytic anaemia, severe pain, coma and eventually death. At that time, it was still an obscure disease, not yet known as TTP.1 It took until 1982 before the ultralarge Von Willebrand Factor (ULVWF) multimers that are typical for TTP were first described.2 In 1998, two research groups described a VWF cleaving protease in TTP.3,4 Finally, in 2001, this proteolytic enzyme was identified as ADAMTS13, a member of the metalloproteinase family.5-7 The VWF cleaving protease ADAMTS13 cleaves the unusually large VWF multimers (VWFMM) into smaller high-molecular weight VWFMM. In TTP patients, an absent or severely reduced ADAMTS13 activity prevents the timely cleavage of ULVWF multimers. These uncleaved multimers induce the adhesion and aggregation of platelets in flowing blood, resulting in the formation of microthrombi (Figure 1).8,9
Figure 1. Thrombotic thrombocytopenic purpura.9
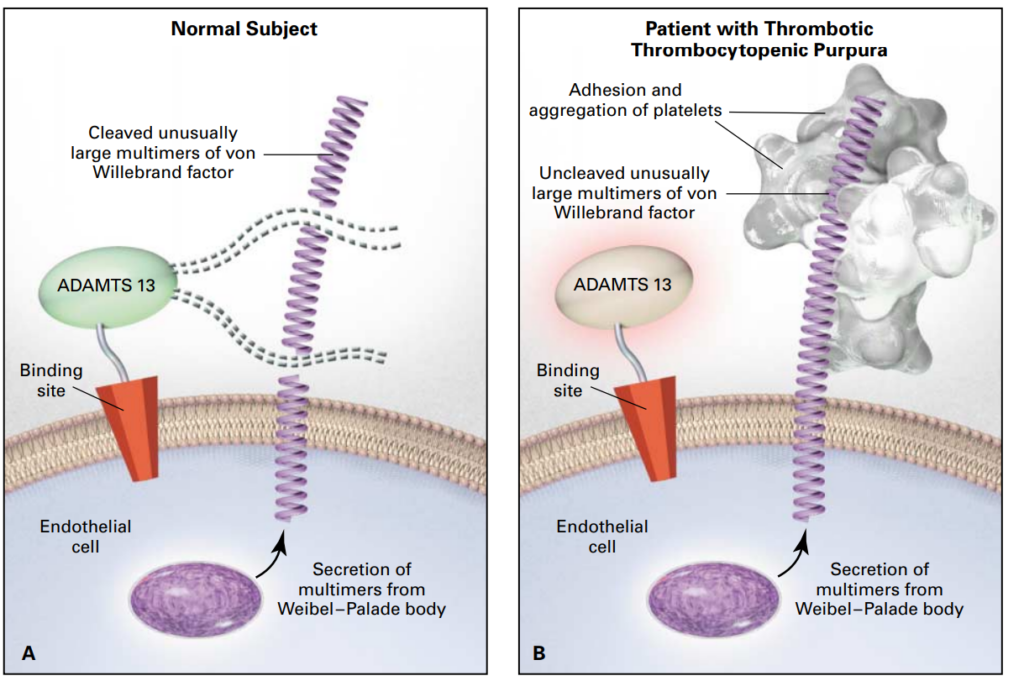
TTP is a rare disease with a prevalence of approximately 10 cases per one million people. The first acute TTP episode usually presents in adults (90% of cases) and the disease is two-fold more frequent in women than in men. ADAMTS13 deficiency is most frequently acquired via ADAMTS13 autoantibodies, but a rare inherited variant has also been described (Upshaw-Schulman syndrome). Despite appropriate therapeutic management, TTP remains a life-threatening disease with a mortality rate of 10-20%.10
By definition, the ADAMTS13 activity is less than 10% in TTP patients. However, this deficiency on its own is it not enough to trigger the clinical symptoms. Other factors that are well established as predisposing factors for acquired TTP include female sex, black ethnicity and obesity. Furthermore, pathophysiological conditions that increase plasma VWF levels (e.g., inflammation, infection or pregnancy) can potentially induce acute episodes of either acquired or inherited TTP. Other still unknown players are suspected to be involved in TTP occurrence. This may include proteins of the ADAMTS13/VWF system or cellular candidates such as platelets or endothelial cells. The historical clinical pentade of TTP includes thrombocytopenia, microangiopathic haemolytic anaemia, neurological symptoms, renal insufficiency and fever. However, all five of these symptoms are only present in approximately 10% of patients with acute TTP.Universal signs of TTP remain severe thrombocytopenia (typically <30 × 109/L) and microangiopathic haemolytic anaemia, characterised by schistocytes on the blood smear. Finally, organ ischemia is common in the brain, heart and kidneys.10
Because TTP belongs to the group of thrombotic microangiopathies (TMAs), making an adequate differential diagnosis from haemolytic uremic syndrome (HUS) caused by the Shiga toxin-producing E. Coli or atypical HUS is of utmost importance. Furthermore, also other TMAs associated with cancer, transplantations or sepsis should be excluded. While patients with TTP can be effectively treated with plasma exchange, this is not the case for patients with other TMAs.10 After excluding other underlying or associated conditions, the TTP diagnosis is based on standard laboratory tests such as the platelet count and creatinine levels. When a platelet count below 30 ×109/L is observed together with a creatinine level below 2.25 mg/dL, TTP should be suspected and ADAMTS13 activity should be measured. However, these results are not always readily available which may cause problems in emergency situations.8 Therefore, the French score or PLASMIC score are often used to get a rapid, preliminary diagnosis. These scores are derived from standard parameters easily available on presentation and offer a comparable and reliable way to identify patients with severe ADAMTS13 deficiency. However, these scores cannot replace the actual ADAMTS13 measurement. Apart from ADAMTS13 activity, also ADAMTS13 antibodies need to be measured to differentiate immune-mediated from congenital TTP, since this has therapeutic consequences.11
The first assay that was described to measure the ADAMTS13 activity is based on a multimer analysis using full length VWF. However, this assay is very time-consuming and difficult to perform in routine practice.12 Subsequently, a fluorescence resonance energy transfer assay (FRET) was developed which uses VWF73, a 73 amino acid residue from D1596 to R1668 of VWF, as a minimal substrate for ADAMTS13. Although this technique is considered to be a reference method, it remains to be very time-consuming.13 The principle of this test was further commercialised in a FRET-based ELISA assay.14 The next step was the development of a monoclonal antibody-based enzyme immunoassay to determine plasma levels of ADAMTS13 activity. This assay is now commercialised under the name Technozyme® ADAMTS13 Activity ELISA.15 The same test principle is also implemented in the automated HemosIL AcuStar ADAMTS13 activity assay with a chemiluminescent readout. This is a rapid assay that is not affected by icterus, lipaemia or plasma turbidity. Based on a large, multicentre, comparative analysis, the HemosIL AcuStar ADAMTS13 Activity assay was found to rapidly provide a result which is largely comparable to the Technozym ADAMTS13 Activity ELISA assay, albeit a slight negative bias.16 In addition to this, there is also a rapid (<1 hour) ADAMTS13 activity test that uses a capture antibody for cleaved VWF73 and a streptavidin gold conjugate. This semi-quantitative assay is however limited by subjective visual interpretation and potential interference by lipids, icterus and haemolysis. Nonetheless, the test is suitable for use in a point of care environment as a screening tool and for the negative exclusion of TTP. When decreased activity is detected, it should be confirmed by a bioassay in an accredited laboratory.14
An International Standard (IS) plasma for ADAMTS13 is available (12/252; NIBSC, South Mimms, UK) and is calibrated for activity and antigen. This plasma should be used as the primary calibrant for commercial calibrants and local assay standards should be traceable to the IS, with results reported as International Units (IU). However, nomenclature, normal range, reporting units and methodology need further standardisation. Furthermore, pre-analytical variables such as sample type and storage should be taken into account. Ideally, samples should be collected before therapy and therapy should already be initiated at clinical suspicion of TTP. According to the International Society on Thrombosis and Haemostasis (ISTH), it is acceptable to have ADAMTS13 test results within 7 days.14
In order to discriminate between congenital and immune-mediated TTP, ADAMTS13 antibodies should be measured. Neutralising antibodies can be demonstrated in mixing tests and Bethesda type assays. These tests detect the antibodies that neutralise the proteolytic activity of ADAMTS13 and the antibodies that increase clearance since many patients have both types of antibodies. Unfortunately, the Bethesda assay has a complex methodology. One Bethesda unit (1BU) is defined as the inhibitor that decreases the residual ADAMTS13 activity to 50% and the inhibitor potency is expressed as BU/ml. An alternative to the Bethesda assay is a chromogenic ELISA. However, with this assay all antibodies are measured, also those without inhibitory activity.14
The management of TTP is based on three important pillars, the inhibition of platelet-VWF interaction, the replenishment of ADAMTS13 levels and immunomodulation to decrease the formation of antibodies. In many cases, treatment therefore consists of a combination of plasma exchange, steroids, caplacizumab and/or rituximab.17 The most recent advancement in TTP treatment is caplacizumab, a nanobody that blocks the binding of the platelet GPIb and the A1 domain of VWF and as such prevents formation of platelet-rich thrombi. In the HERCULES trial, treatment with caplacizumab resulted in a faster normalisation of platelet count, a lower incidence of TTP related death, a lower incidence of recurrence during the trial and a lower incidence of thrombotic events during treatment period. Importantly, caplacizumab should be initiated as soon as possible, along with plasma exchange.18
In settings with a timely access to plasma ADAMTS13 activity testing and for patients with a high clinical suspicion (≥90% pretest probability) of iTTP, the ISTH recommends to acquire a plasma sample for ADAMTS13 testing before an initiation of plasma exchange or the use of other blood products. Early administration of caplacizumab should be considered before test results become available. When the result of plasma a ADAMTS13 activity assessment becomes available, caplacizumab should be continued if the ADAMTS13 activity is less than 10 IU/dL (or <10% of normal) and stopped if ADAMTS13 activity is >20 IU/dL (or >20% of normal). Clinical judgement is required for continuing or stopping caplacizumab in case of an equivocal result. For patients with intermediate or low clinical suspicion of iTTP based on pretests, caplacizumab should not be initiated until the result of a plasma ADAMTS13 activity test becomes available.19
Furthermore, ADAMTS13 activity not only needs to be measured at diagnosis but should also be monitored during treatment in order to timely detect exacerbation or TTP relapse. In practice, the ADAMTS13 activity should be measured weekly until it reaches at least 50%. Importantly, the same ADAMTS13 assay should be used within the same patient in order to exclude variability amongst the different assays. Once ADAMTS13 activity reaches 50%, it should be measured every three months for the first three years, every six months for the next two years and annually thereafter. If the ADAMTS13 activity again decreases to 20-49%, more frequent monitoring should be done and therapeutic adjustments should be considered. In addition, also platelet counts should be measured.20
Finally, as relapses are common in TTP, there is a lot of interest in new biomarkers. For example, an open conformation of ADAMTS13 was found to be a hallmark of acute acquired TTP. Therefore, monitoring the open ADAMTS13 may allow early detection of subclinical TTP and initiation of prompt therapy. In addition, it can be an aid in diagnosis (especially for patients with borderline ADAMTS13 activity between 10-20%) and follow-up of TTP patients. However, to date we are still awaiting the commercial availability of this assay.21
References
